


21 мая 2024 Финам Бурмистров Геннадий
В этой статье поговорим о логике оценки биотехнологических компаний в принципе и о тех компаниях, у которых близки рассмотрения FDA в мае-июне 2024.
На первом этапе при изучении компании биотехнологического сектора стоит обратить внимание на то, какие клинические исследования она проводит, и на какой стадии они находятся.
В клинических исследованиях разрабатываемые молекулы терапии тестируются на людях. Любое такое исследование разделено на три этапа. Прежде всего компания должна подать заявку в управление по санитарному надзору за качеством пищевых продуктов и медикаментов (оно же FDA), на начало клинических исследований нового препарата и получить от него одобрение. Это происходит только после того, как проведено большое количество экспериментов и собрано обширное количество данных в опытах in vitro, то есть тех, которые проводятся «в пробирке» — вне живого организма. Если FDA посчитает убедительными данные, собранные в доклинических испытаниях, то оно выдаёт компании одобрение на проведение клинических испытаний, которые делятся на первую, вторую и третью фазы.

В первой фазе клинических испытаний могут участвовать как здоровые добровольцы, так и пациенты с определенным заболеванием. Во время I фазы изучается насколько хорошо переносится исследуемый препарат, оценивается безопасность новой молекулы. Различные дозы лекарства тестируются на узкой популяции добровольцев. Вся информация обрабатывается, а после того, как в ходе исследований врачи изучили фармакодинамику, фармакокинетику и предварительную безопасность исследуемого лекарственного препарата, подаётся запрос в FDA на разрешение проведения второй фазы.

Во второй фазе участвуют пациенты, страдающие патологией, для лечения которой предназначен исследуемый препарат. Основная цель клинических исследований второй фазы - найти оптимальный уровень дозировки и подобрать схему приёма препарата для следующей, третьей фазы.
Дозы препарата, которые получают пациенты на данном этапе, зачастую ниже, чем самые высокие дозы, которые принимали участники фазы I.

Третья фаза оценивает эффективность препарата по сравнению с другими, уже действующими стандартами лечения болезни. В ней также участвуют пациенты, страдающие патологией, но исследования проводятся на гораздо большей популяции больных людей, чем во второй фазе.
По завершению каждой стадии компания, как правило публикует результаты исследований в общедоступных источниках, например, в пресс-релизах, презентациях или же на различных медицинских конференциях, касающихся исследуемых заболеваний. В момент освещения результатов тестирования разрабатываемой молекулы, когда данные предоставляются на обозрение общественности, мы можем наблюдать существенные движения в котировках цены акций компании.
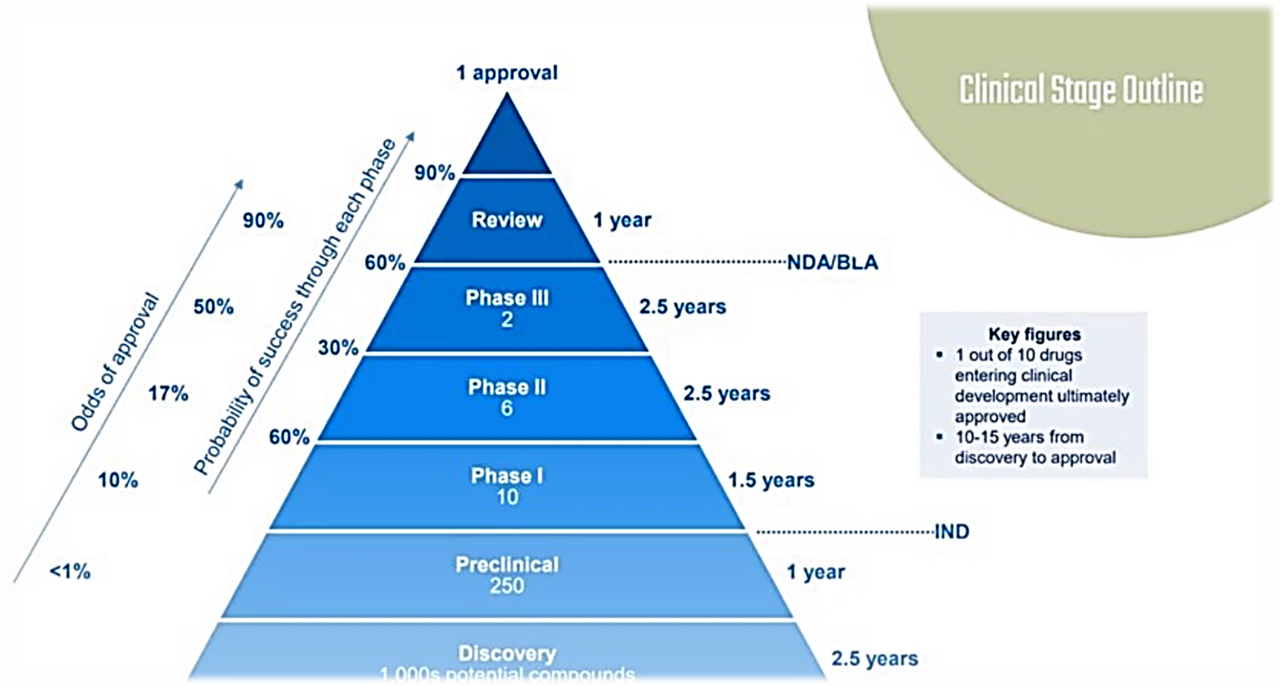
На данном слайде представлена так называемая воронка принятия новых медицинских препаратов. Как вы видите, от момента открытия молекулы до момента принятия её в качестве лекарства в среднем может пройти от 10 до 15 лет.
В пирамиде указаны различные данные, такие как сроки проведения каждого этапа при продвижении нового препарата к принятию, процент одобрения при переходе на новый уровень и так далее. Так, например, средние сроки проведения доклинических испытаний составляют 1 год, и только 4% исследуемых молекул получают одобрение от FDA, чтобы начать их испытание на людях.
Вторая фаза в среднем длится 2,5 года, и только 30% потенциальных лекарств смогут перейти к испытаниям на более широкой популяции больных.
Как вы видите, вероятность того, что новая разработка переродится в одобренный препарат крайне мала. Из 1000 потенциальных кандидатов, по статистике, только один будет коммерциализирован. Именно поэтому положительные или отрицательные результаты исследования на каждой фазе, а следовательно, и продвижение её кандидата к заветному одобрению так сильно влияют на создание стоимости компании.
Давайте теперь немного поговорим о регулирующих органах, которые занимаются оценкой безопасности и эффективности исследований в медицинской сфере. Это FDA, в США и EMA в Европе. Есть также китайская регулирующая организация, которая является довольно авторитетной.
На что в первую очередь смотрят FDA и EMA, когда им на рассмотрение поступает новая молекула?
- на безопасность
- на эффективность
- и на текущие стандарты лечения
Если новое лекарство является более безопасным чем текущие стандартные лечения, то есть имеет меньше побочных эффектов для пациента, или эти побочные эффекты более низкой степени, то регуляторы приступают к рассмотрению его эффективности.
Также может оказаться, что у болезни, против которой направлена исследуемая терапия, отсутствуют методы лечения, тогда порог безопасности будет существенно снижен. Так, например, могут быть приняты к рассмотрению лекарства с очень серьезными побочными эффектами, даже 4 степени тяжести, в случае если заболевание является смертельным и для него пока нет действующих препаратов. Такой же подход применяется в эффективности. Достаточным для одобрения порогом необходимой эффективности является превосходство в этом плане над терапией, которая в данный момент является стандартом лечения. Если в данный момент нет ничего, что помогает пациентам при заболевании, этот порог может быть довольно низким. Если же в данный момент на рынке существует много одобренных лекарств для лечения болезни, тогда кандидат может столкнуться с очень высокими требованиями по эффективности.
Регулирующие органы могут присваивать определённые статусы молекулам, которые посчитали особо полезными или особо эффективными, или же, если существует неудовлетворённая медицинская потребность по болезни, на борьбу с которой направлена разработка последней. Эти статусы созданы с целью помочь потенциальным препаратам как можно быстрее пройти через пирамиду принятия, рассмотренную выше.

Fast Track — это статус, предназначенный для облегчения разработки и ускорения обзора лекарств для лечения серьезных состояний и удовлетворения неудовлетворенных медицинских потребностей. Цель его состоит в том, чтобы как можно раньше доставить пациенту важные новые лекарства. Определение серьезности состояния является вопросом относительным, но обычно основывается на том, будет ли лекарство влиять на такие факторы, как выживаемость, повседневное функционирование или вероятность того, что состояние, если его не лечить, будет прогрессировать от менее тяжелого состояния к более серьезному.
СПИД, болезнь Альцгеймера, сердечная недостаточность и рак - очевидные примеры серьезных заболеваний. Также такие заболевания, как эпилепсия, депрессия и диабет, также считаются серьезными состояниями.
Заполнение неудовлетворенной медицинской потребности определяется как предоставление терапии там, где ее не существует, или обеспечение терапии, которая потенциально может быть лучше, чем доступная терапия. Любое лекарство, разрабатываемое для лечения или предотвращения состояния без текущей терапии, очевидно, направлено на неудовлетворенную потребность.
Лекарство, получившее обозначение Fast Track, получает следующие преимущества:
- Более частые встречи с FDA для обсуждения плана разработки препарата и обеспечения сбора соответствующих данных, необходимых для поддержки утверждения препарата.
- Более частые письменные сообщения от FDA о таких вещах, как дизайн предлагаемых клинических испытаний и использование биомаркеров.
- Право на ускоренное одобрение и приоритетную проверку при соблюдении соответствующих критериев
- Постоянное рассмотрение, что означает, что фармацевтическая компания может отправить на рассмотрение некоторые разделы, которые уже заполнены из своей заявки на биологическую лицензию или заявку на новый лекарственный препарат на рассмотрение FDA вместо того, чтобы ждать, пока каждый раздел NDA будет заполнен
В обычных условиях проверка BLA или NDA не начинается до тех пор, пока фармацевтическая компания не представит полностью заполненную заявку в FDA.

Прорывная терапия — это статус, предназначенный для ускорения разработки и обзора лекарств, предназначенных для лечения серьезного состояния. Назначается в том случае, если предварительные клинические данные указывают на то, что лекарство может продемонстрировать значительное улучшение по сравнению с доступной терапией по клинически значимым конечным точкам. Определение того, является ли улучшение по сравнению с доступной терапией значительным, является предметом суждения и зависит как от величины лечебного эффекта, который может включать продолжительность эффекта, так и от важности наблюдаемого клинического результата.
В целом предварительные клинические данные должны показать явное преимущество перед доступной терапией.
Препарат, получивший статус прорывной терапии, получает следующие преимущества:
- Все плюсы обозначения Fast Track
- Интенсивное руководство по эффективной программе разработки лекарств, начиная с фазы 1
- Доступ к обсуждениям новой терапии с участием старших менеджеров FDA

Ускоренное одобрение – сокращает время рассмотрения на одобрение препарата. Этот статус позволяет произвести одобрение лекарств для серьезных состояний, удовлетворяющих неудовлетворенную медицинскую потребность, на основе суррогатной конечной точки.
При изучении нового лекарства иногда может потребоваться много лет, чтобы узнать, действительно ли лекарство оказывает реальное влияние на то, как пациент выживает, чувствует или функционирует.
Принимая во внимание тот факт, что для измерения предполагаемой клинической пользы препарата может потребоваться длительный период времени. В 1992 г. FDA ввело правила ускоренного утверждения по суррогатной конечно точке.
Суррогатная конечная точка, используемая для ускоренного утверждения, является маркером - лабораторным измерением, рентгенографическим изображением, физическим признаком или другим показателем, который, как считается, предсказывает клиническую пользу, но сам по себе не является мерой клинической пользы.
Использование суррогатных или промежуточных клинических конечных точек может сэкономить драгоценное время в процессе утверждения лекарства. Например, вместо того чтобы ждать, чтобы узнать, действительно ли лекарство увеличивает выживаемость онкологических больных, FDA может одобрить лекарство на основании доказательств того, что молекула уменьшает опухоли, поскольку уменьшение размеров опухоли может с достаточной вероятностью предсказывать реальную клиническую пользу.
Статус терапии орфанного заболевания, то есть заболевания, которое затрагивает небольшую часть населения, даёт преимущества в плане поддержки кандидата от государства, например более длительный патент по сравнению с другими препаратами.

Статус приоритетного обзора сокращает время рассмотрения FDA заявки на лекарство, поданное компанией после прохождения 3 фазы испытаний со стандартных 10 до ускоренных 6 месяцев.
Перед утверждением каждое лекарство, продаваемое в Соединенных Штатах, должно пройти детальную проверку FDA. Назначение приоритетной проверки направляет общее внимание и ресурсы агентства на оценку заявок на лекарственные препараты, которые в случае одобрения значительно улучшат безопасность или эффективность лечения, диагностики или профилактики серьезных состояний по сравнению со стандартными заявками.
Значительное улучшение можно продемонстрировать на следующих примерах:
- доказательства повышенной эффективности лечения, профилактики или диагностики состояния;
- устранение или существенное уменьшение реакции на лекарственное средство, ограничивающую лечение;
- задокументированное улучшение соблюдения пациентом режима лечения, которое, как ожидается, приведет к улучшению серьезных исходов;
- доказательства безопасности и эффективности в новой подгруппе населения.
Что нужно сделать для предположения об объеме рынка?
Оценить количество пациентов, страдающих от заболевания, на которое нацелена терапия. Такие данные публикуют ВОЗ и Центры США по контролю и профилактике заболеваний (CDC).
Оценить конкуренцию. На сайте CDC часто публикуются данные по другим терапиям для конкретного заболевания. На их основе можно предположить, на какую долю рынка претендует препарат.
Для определения перспектив препарата следует определять целевой рынок и частоту возникновения заболевания на нем. Некоторые заболевания обладают различной частотой возникновения в разных этнических группах. Например, серповидно-клеточная анемия распространена среди народов Африки, Индии, Средиземноморья и Средней Азии. Прогнозировать стоимость препарата можно методом сравнения аналогов или оценкой стоимости выгод, полученных пациентом от лечения, считает эксперт. Подобные расчеты публикуют аналитические агентства, такие как Institute for Clinical and Economic Review (ICER).
Определение объема рынка — это сложная и творческая задача, требующая глубокого понимания отрасли и действий конкурентов. Эксперт обращает внимание, что зачастую в перспективных нишах уже идет ожесточенная конкуренция между десятком биотехов.
В инновационных разработках главное не рынок лечения конкретного заболевания, а валидация эффективности способа действия препарата. Например, если небольшая компания, вроде Allogene Therapeutics, докажет высокую эффективность хотя бы одного препарата, для рынка это станет доказательством работы всего подхода, который можно быстро масштабировать для лечения схожих заболеваний.
Разработка лекарств стоит дорого. Стоимость разработки успешного препарата с учетом множества неудачных попыток превышает $2,5 млрд и это довольно приблизительная, даже несколько заниженная цифра. Разработка лекарств требует огромного капитала — практически невозможно запустить фармкомпанию без инвесторов.
У большинства биотехов нет доходов и прибыли. Фактически денежные потоки до одобрения FDA препарата-кандидата будут отрицательными. Прибыли нет у двух третей компаний, входящих в состав индекса биотехов NASDAQ Biotech Index (NBI), но у большинства из них капитализация превышает $1 млрд.
По этой причине стандартные мультипликаторы вроде EV/ EBITDA или P/E неприменимы для оценки биотехов. Существуют альтернативные коэффициенты, например отношение EV к инвестированным в НИОКР средства — по сути, это оценка, основанная на затратах.
Сравнение с другими биотехами часто также бесполезно. Многие компании в секторе индивидуальны, и их перспективы строятся на идее, защищаемой патентом. Из-за этого просто экстраполировать исторические доходы уже одобренных препаратов бессмысленно.
Часто после нескольких лет производства даже успешного препарата доходы от него падают. Это связано с истечением срока действия патента и последующей конкуренцией со стороны копий или дженериков. В США стандартный срок патентной защиты составляет 20 лет. Однако новые лекарства обычно запатентовываются на ранних стадиях — во время испытаний на животных на доклинической стадии. С учетом того, что до одобрения препарата может пройти восемь — десять лет, фактически доход защищен только десять лет.
На первом этапе при изучении компании биотехнологического сектора стоит обратить внимание на то, какие клинические исследования она проводит, и на какой стадии они находятся.
В клинических исследованиях разрабатываемые молекулы терапии тестируются на людях. Любое такое исследование разделено на три этапа. Прежде всего компания должна подать заявку в управление по санитарному надзору за качеством пищевых продуктов и медикаментов (оно же FDA), на начало клинических исследований нового препарата и получить от него одобрение. Это происходит только после того, как проведено большое количество экспериментов и собрано обширное количество данных в опытах in vitro, то есть тех, которые проводятся «в пробирке» — вне живого организма. Если FDA посчитает убедительными данные, собранные в доклинических испытаниях, то оно выдаёт компании одобрение на проведение клинических испытаний, которые делятся на первую, вторую и третью фазы.
В первой фазе клинических испытаний могут участвовать как здоровые добровольцы, так и пациенты с определенным заболеванием. Во время I фазы изучается насколько хорошо переносится исследуемый препарат, оценивается безопасность новой молекулы. Различные дозы лекарства тестируются на узкой популяции добровольцев. Вся информация обрабатывается, а после того, как в ходе исследований врачи изучили фармакодинамику, фармакокинетику и предварительную безопасность исследуемого лекарственного препарата, подаётся запрос в FDA на разрешение проведения второй фазы.
Во второй фазе участвуют пациенты, страдающие патологией, для лечения которой предназначен исследуемый препарат. Основная цель клинических исследований второй фазы - найти оптимальный уровень дозировки и подобрать схему приёма препарата для следующей, третьей фазы.
Дозы препарата, которые получают пациенты на данном этапе, зачастую ниже, чем самые высокие дозы, которые принимали участники фазы I.
Третья фаза оценивает эффективность препарата по сравнению с другими, уже действующими стандартами лечения болезни. В ней также участвуют пациенты, страдающие патологией, но исследования проводятся на гораздо большей популяции больных людей, чем во второй фазе.
По завершению каждой стадии компания, как правило публикует результаты исследований в общедоступных источниках, например, в пресс-релизах, презентациях или же на различных медицинских конференциях, касающихся исследуемых заболеваний. В момент освещения результатов тестирования разрабатываемой молекулы, когда данные предоставляются на обозрение общественности, мы можем наблюдать существенные движения в котировках цены акций компании.
На данном слайде представлена так называемая воронка принятия новых медицинских препаратов. Как вы видите, от момента открытия молекулы до момента принятия её в качестве лекарства в среднем может пройти от 10 до 15 лет.
В пирамиде указаны различные данные, такие как сроки проведения каждого этапа при продвижении нового препарата к принятию, процент одобрения при переходе на новый уровень и так далее. Так, например, средние сроки проведения доклинических испытаний составляют 1 год, и только 4% исследуемых молекул получают одобрение от FDA, чтобы начать их испытание на людях.
Вторая фаза в среднем длится 2,5 года, и только 30% потенциальных лекарств смогут перейти к испытаниям на более широкой популяции больных.
Как вы видите, вероятность того, что новая разработка переродится в одобренный препарат крайне мала. Из 1000 потенциальных кандидатов, по статистике, только один будет коммерциализирован. Именно поэтому положительные или отрицательные результаты исследования на каждой фазе, а следовательно, и продвижение её кандидата к заветному одобрению так сильно влияют на создание стоимости компании.
Давайте теперь немного поговорим о регулирующих органах, которые занимаются оценкой безопасности и эффективности исследований в медицинской сфере. Это FDA, в США и EMA в Европе. Есть также китайская регулирующая организация, которая является довольно авторитетной.
На что в первую очередь смотрят FDA и EMA, когда им на рассмотрение поступает новая молекула?
- на безопасность
- на эффективность
- и на текущие стандарты лечения
Если новое лекарство является более безопасным чем текущие стандартные лечения, то есть имеет меньше побочных эффектов для пациента, или эти побочные эффекты более низкой степени, то регуляторы приступают к рассмотрению его эффективности.
Также может оказаться, что у болезни, против которой направлена исследуемая терапия, отсутствуют методы лечения, тогда порог безопасности будет существенно снижен. Так, например, могут быть приняты к рассмотрению лекарства с очень серьезными побочными эффектами, даже 4 степени тяжести, в случае если заболевание является смертельным и для него пока нет действующих препаратов. Такой же подход применяется в эффективности. Достаточным для одобрения порогом необходимой эффективности является превосходство в этом плане над терапией, которая в данный момент является стандартом лечения. Если в данный момент нет ничего, что помогает пациентам при заболевании, этот порог может быть довольно низким. Если же в данный момент на рынке существует много одобренных лекарств для лечения болезни, тогда кандидат может столкнуться с очень высокими требованиями по эффективности.
Регулирующие органы могут присваивать определённые статусы молекулам, которые посчитали особо полезными или особо эффективными, или же, если существует неудовлетворённая медицинская потребность по болезни, на борьбу с которой направлена разработка последней. Эти статусы созданы с целью помочь потенциальным препаратам как можно быстрее пройти через пирамиду принятия, рассмотренную выше.
Fast Track — это статус, предназначенный для облегчения разработки и ускорения обзора лекарств для лечения серьезных состояний и удовлетворения неудовлетворенных медицинских потребностей. Цель его состоит в том, чтобы как можно раньше доставить пациенту важные новые лекарства. Определение серьезности состояния является вопросом относительным, но обычно основывается на том, будет ли лекарство влиять на такие факторы, как выживаемость, повседневное функционирование или вероятность того, что состояние, если его не лечить, будет прогрессировать от менее тяжелого состояния к более серьезному.
СПИД, болезнь Альцгеймера, сердечная недостаточность и рак - очевидные примеры серьезных заболеваний. Также такие заболевания, как эпилепсия, депрессия и диабет, также считаются серьезными состояниями.
Заполнение неудовлетворенной медицинской потребности определяется как предоставление терапии там, где ее не существует, или обеспечение терапии, которая потенциально может быть лучше, чем доступная терапия. Любое лекарство, разрабатываемое для лечения или предотвращения состояния без текущей терапии, очевидно, направлено на неудовлетворенную потребность.
Лекарство, получившее обозначение Fast Track, получает следующие преимущества:
- Более частые встречи с FDA для обсуждения плана разработки препарата и обеспечения сбора соответствующих данных, необходимых для поддержки утверждения препарата.
- Более частые письменные сообщения от FDA о таких вещах, как дизайн предлагаемых клинических испытаний и использование биомаркеров.
- Право на ускоренное одобрение и приоритетную проверку при соблюдении соответствующих критериев
- Постоянное рассмотрение, что означает, что фармацевтическая компания может отправить на рассмотрение некоторые разделы, которые уже заполнены из своей заявки на биологическую лицензию или заявку на новый лекарственный препарат на рассмотрение FDA вместо того, чтобы ждать, пока каждый раздел NDA будет заполнен
В обычных условиях проверка BLA или NDA не начинается до тех пор, пока фармацевтическая компания не представит полностью заполненную заявку в FDA.
Прорывная терапия — это статус, предназначенный для ускорения разработки и обзора лекарств, предназначенных для лечения серьезного состояния. Назначается в том случае, если предварительные клинические данные указывают на то, что лекарство может продемонстрировать значительное улучшение по сравнению с доступной терапией по клинически значимым конечным точкам. Определение того, является ли улучшение по сравнению с доступной терапией значительным, является предметом суждения и зависит как от величины лечебного эффекта, который может включать продолжительность эффекта, так и от важности наблюдаемого клинического результата.
В целом предварительные клинические данные должны показать явное преимущество перед доступной терапией.
Препарат, получивший статус прорывной терапии, получает следующие преимущества:
- Все плюсы обозначения Fast Track
- Интенсивное руководство по эффективной программе разработки лекарств, начиная с фазы 1
- Доступ к обсуждениям новой терапии с участием старших менеджеров FDA
Ускоренное одобрение – сокращает время рассмотрения на одобрение препарата. Этот статус позволяет произвести одобрение лекарств для серьезных состояний, удовлетворяющих неудовлетворенную медицинскую потребность, на основе суррогатной конечной точки.
При изучении нового лекарства иногда может потребоваться много лет, чтобы узнать, действительно ли лекарство оказывает реальное влияние на то, как пациент выживает, чувствует или функционирует.
Принимая во внимание тот факт, что для измерения предполагаемой клинической пользы препарата может потребоваться длительный период времени. В 1992 г. FDA ввело правила ускоренного утверждения по суррогатной конечно точке.
Суррогатная конечная точка, используемая для ускоренного утверждения, является маркером - лабораторным измерением, рентгенографическим изображением, физическим признаком или другим показателем, который, как считается, предсказывает клиническую пользу, но сам по себе не является мерой клинической пользы.
Использование суррогатных или промежуточных клинических конечных точек может сэкономить драгоценное время в процессе утверждения лекарства. Например, вместо того чтобы ждать, чтобы узнать, действительно ли лекарство увеличивает выживаемость онкологических больных, FDA может одобрить лекарство на основании доказательств того, что молекула уменьшает опухоли, поскольку уменьшение размеров опухоли может с достаточной вероятностью предсказывать реальную клиническую пользу.
Статус терапии орфанного заболевания, то есть заболевания, которое затрагивает небольшую часть населения, даёт преимущества в плане поддержки кандидата от государства, например более длительный патент по сравнению с другими препаратами.
Статус приоритетного обзора сокращает время рассмотрения FDA заявки на лекарство, поданное компанией после прохождения 3 фазы испытаний со стандартных 10 до ускоренных 6 месяцев.
Перед утверждением каждое лекарство, продаваемое в Соединенных Штатах, должно пройти детальную проверку FDA. Назначение приоритетной проверки направляет общее внимание и ресурсы агентства на оценку заявок на лекарственные препараты, которые в случае одобрения значительно улучшат безопасность или эффективность лечения, диагностики или профилактики серьезных состояний по сравнению со стандартными заявками.
Значительное улучшение можно продемонстрировать на следующих примерах:
- доказательства повышенной эффективности лечения, профилактики или диагностики состояния;
- устранение или существенное уменьшение реакции на лекарственное средство, ограничивающую лечение;
- задокументированное улучшение соблюдения пациентом режима лечения, которое, как ожидается, приведет к улучшению серьезных исходов;
- доказательства безопасности и эффективности в новой подгруппе населения.
Что нужно сделать для предположения об объеме рынка?
Оценить количество пациентов, страдающих от заболевания, на которое нацелена терапия. Такие данные публикуют ВОЗ и Центры США по контролю и профилактике заболеваний (CDC).
Оценить конкуренцию. На сайте CDC часто публикуются данные по другим терапиям для конкретного заболевания. На их основе можно предположить, на какую долю рынка претендует препарат.
Для определения перспектив препарата следует определять целевой рынок и частоту возникновения заболевания на нем. Некоторые заболевания обладают различной частотой возникновения в разных этнических группах. Например, серповидно-клеточная анемия распространена среди народов Африки, Индии, Средиземноморья и Средней Азии. Прогнозировать стоимость препарата можно методом сравнения аналогов или оценкой стоимости выгод, полученных пациентом от лечения, считает эксперт. Подобные расчеты публикуют аналитические агентства, такие как Institute for Clinical and Economic Review (ICER).
Определение объема рынка — это сложная и творческая задача, требующая глубокого понимания отрасли и действий конкурентов. Эксперт обращает внимание, что зачастую в перспективных нишах уже идет ожесточенная конкуренция между десятком биотехов.
В инновационных разработках главное не рынок лечения конкретного заболевания, а валидация эффективности способа действия препарата. Например, если небольшая компания, вроде Allogene Therapeutics, докажет высокую эффективность хотя бы одного препарата, для рынка это станет доказательством работы всего подхода, который можно быстро масштабировать для лечения схожих заболеваний.
Разработка лекарств стоит дорого. Стоимость разработки успешного препарата с учетом множества неудачных попыток превышает $2,5 млрд и это довольно приблизительная, даже несколько заниженная цифра. Разработка лекарств требует огромного капитала — практически невозможно запустить фармкомпанию без инвесторов.
У большинства биотехов нет доходов и прибыли. Фактически денежные потоки до одобрения FDA препарата-кандидата будут отрицательными. Прибыли нет у двух третей компаний, входящих в состав индекса биотехов NASDAQ Biotech Index (NBI), но у большинства из них капитализация превышает $1 млрд.
По этой причине стандартные мультипликаторы вроде EV/ EBITDA или P/E неприменимы для оценки биотехов. Существуют альтернативные коэффициенты, например отношение EV к инвестированным в НИОКР средства — по сути, это оценка, основанная на затратах.
Сравнение с другими биотехами часто также бесполезно. Многие компании в секторе индивидуальны, и их перспективы строятся на идее, защищаемой патентом. Из-за этого просто экстраполировать исторические доходы уже одобренных препаратов бессмысленно.
Часто после нескольких лет производства даже успешного препарата доходы от него падают. Это связано с истечением срока действия патента и последующей конкуренцией со стороны копий или дженериков. В США стандартный срок патентной защиты составляет 20 лет. Однако новые лекарства обычно запатентовываются на ранних стадиях — во время испытаний на животных на доклинической стадии. С учетом того, что до одобрения препарата может пройти восемь — десять лет, фактически доход защищен только десять лет.
Не является индивидуальной инвестиционной рекомендацией | При копировании ссылка обязательна | Нашли ошибку - выделить и нажать Ctrl+Enter | Жалоба